 ¿Qué es demasiado pequeño para ver a simple vista, fabricado por la mitad de la población en lotes de millones y con una oferta alarmante? La respuesta, según algunos científicos, es el esperma. Específicamente, a los investigadores les preocupa que los hombres en Occidente hayan estado produciendo cada vez menos espermatozoides desde la década de 1970, una disminución que, según ellos, no muestra signos de detenerse. Al ritmo actual, dicen, estos hombres podrían ser infértiles en 2045. Pero estas cifras deberían hacernos pensar. La idea de que el esperma de los hombres en los países occidentales está a punto de colapsar es, en una palabra, extraordinaria. Los datos no lo respaldan.
El temor a una disminución en el recuento de espermatozoides es potente y de gran alcance. Ha sido expresado por todos, desde ambientalistas como Erin Brockovich hasta supremacistas blancos y sus portavoces de los principales medios de comunicación como Tucker Carlson. El gran atractivo de esta noción es posible porque las supuestas causas de un declive abarcan toda la gama material e ideológica. Incluyen sustancias químicas que se encuentran en productos domésticos comunes, así como estilos de vida urbanos modernos en los que los hombres blancos son físicamente sedentarios y se ven obligados a compartir el poder con personas de géneros y etnias en constante diversidad. Vale la pena echar un vistazo más de cerca a la evidencia antes de acudir al mitin de "salvar el esperma". La ronda más reciente de predicciones apocalípticas fue provocada por un influyente artículo de 2017 que recopiló datos de recuento de espermatozoides de estudios publicados entre 1973 y 2011. Las cifras de los EE. UU., Canadá, Europa occidental, Australia y Nueva Zelanda, es decir, países ricos en su mayoría blancos, fueron agrupados en la categoría "occidental" y los datos restantes se agruparon en la categoría "otros". Si bien no había datos suficientes para sacar conclusiones sobre "otros" hombres, los autores encontraron que el recuento de espermatozoides de la población promedio entre los hombres en la categoría "occidental" había disminuido en más del 50% desde 1973. Analizamos los datos y descubrimos que el veredicto apocalíptico de la desaparición de los espermatozoides está lejos de ser la única interpretación plausible de lo que está sucediendo aquí. Los autores asumen que los altos recuentos de espermatozoides de los hombres en las naciones "occidentales" en la década de 1970 representan la norma. Esta suposición comete el error pernicioso pero demasiado común de tratar a los hombres de las naciones prósperas y de mayoría blanca como el estándar con el que todos los demás deberían ser comparados. También da por sentado que, cuando se trata de espermatozoides, cuanto más, mejor. La evidencia disponible no respalda esta asociación: la fertilidad masculina no escala proporcionalmente con el recuento de espermatozoides. Algunos hombres con un recuento bajo de espermatozoides pueden ser muy fértiles, mientras que otros que rebosan de espermatozoides pueden tener dificultades para concebir. Agregue a esto el hecho bien conocido de que el recuento de espermatozoides es muy sensible al contexto (a la ropa interior ajustada, el ejercicio, un baño caliente e incluso a la temporada) y es fácil ver cómo una sola medida del recuento de espermatozoides es un indicador poco confiable de fertilidad. . También está la pregunta de qué está causando la disminución en el recuento de espermatozoides. Si nos tomamos en serio la idea de que los contaminantes ambientales son hostiles a la producción de esperma, esperaríamos ver los descensos más drásticos entre los hombres que viven en los entornos más contaminados. Está bien establecido que los pobres del mundo, es decir, los que viven predominantemente (pero no exclusivamente) en los "otros" países, soportan la mayor carga de contaminación ambiental. Sin embargo, los autores y los medios de comunicación se han lanzado a enmarcar la crisis como una que enfrenta a los hombres "occidentales"; lo que se ignora es el hecho de que los datos del estudio eran insuficientes para sacar conclusiones para los hombres en la categoría "otros". El mismo uso de las categorías "occidental" y "otros" tiene poco sentido científico e importa matices raciales peligrosos. Los hombres migran con frecuencia entre naciones "occidentales" y "otras", lo que hace que los países sean un pobre indicador de los entornos que podrían afectar el esperma de cualquier hombre. Y las condiciones varían ampliamente dentro de las naciones, especialmente en las grandes y heterogéneas como Estados Unidos o Brasil. Saber qué pasaporte lleva un hombre le dice poco sobre los contaminantes u otros posibles factores reductores de esperma que puede haber encontrado. Aparte de todo esto, estos hallazgos aparentemente alarmantes pueden simplemente reflejar una variación normal. Esto no tendría precedentes: los estudios han documentado altibajos naturales en los niveles de hormonas reproductivas como la testosterona y la progesterona, sin impacto en la fertilidad. ¿Podrían variar los recuentos de espermatozoides de la misma manera? Los investigadores ni siquiera consideran esta posibilidad. La lección de la investigación sobre la disminución de los espermatozoides no es que nos enfrentemos a una extinción humana inminente (al menos no por razones relacionadas con los espermatozoides). Más bien, es el hecho más banal pero exacto de que hay mucho que desconocemos sobre la relación entre la salud reproductiva de los hombres y la contaminación ambiental. Este punto ciego es a lo que deberíamos prestar atención. Una larga y sexista historia de científicos que se centran celosamente en la reproducción de las mujeres ha llevado a los investigadores a descuidar la fertilidad de los hombres. El legado del cabildeo de la industria química y la investigación financiada por la industria distorsiona nuestro conocimiento de los efectos de la exposición a los plásticos en la salud humana. Y una historia racista de tratar los cuerpos masculinos blancos ricos como la norma de la especie nos prepara para ignorar a la mayoría de la población mundial. Frente a toda esta incertidumbre y ofuscación, lo que necesitamos es una mejor ciencia: instituciones científicas libres de la influencia corporativa y diversos investigadores capacitados para desenterrar supuestos racistas y sexistas ocultos. Nuestro incumplimiento de estos estándares es la verdadera razón del pánico. Source:https://www.theguardian.com/commentisfree/2021/jun/07/scare-stories-falling-sperm-counts-male-infertility-science
0 Comentarios
Daniel Beck 1, Millissia Ben Maamar 1, Michael K Skinner 2 ANTECEDENTES Durante las últimas dos décadas, numerosos estudios han demostrado una forma de herencia no genética denominada herencia transgeneracional epigenética que está mediada por alteraciones de la línea germinal en los procesos epigenéticos [1-3]. Una de las primeras observaciones involucró la vinclozolina, un tóxico agrícola ambiental, que es uno de los fungicidas agrícolas más utilizados, para inducir la herencia transgeneracional epigenética de la patología testicular y alteraciones de la metilación del ADN [1]. Observaciones similares con una amplia variedad de tóxicos ambientales, desde dioxinas hasta DDT (dicloro-difenil-tricloroetano), han identificado impactos de herencia epigenética similares en una variedad de enfermedades diferentes [3-5]. Las manifestaciones fenotípicas transgeneracionales de vinclozolina y DDT incluyen la inducción de patología testicular, prostática, renal y ovárica, así como obesidad [3]. Una observación temprana en ratones identificó un impacto traumático inducido por el estrés en la herencia transgeneracional epigenética de las anomalías conductuales [6, 7]. Curiosamente, la inyección de óvulos con el ncRNA de espermatozoides masculinos individuales estresados promovió los mismos fenotipos transgeneracionales [6]. Estudios posteriores han apoyado el papel de la metilación del ADN o del ARNnc en la herencia transgeneracional epigenética mediada por la línea germinal [3, 8]. Se ha demostrado que este fenómeno de herencia transgeneracional epigenética es inducido por sustancias químicas ambientales, nutrición, estrés y anomalías traumáticas en roedores y humanos [3, 7, 9], así como por una amplia variedad de tensiones ambientales en las plantas [10, 11], insectos [12, 13], gusanos [14], peces [15-17], aves [18, 19] y una variedad de mamíferos como cerdos y humanos [20-22]. Se han observado varios impactos fisiológicos que incluyen patologías en el cerebro, los órganos reproductores, los riñones, la inmunidad, la obesidad y la infertilidad [1-3]. El fenómeno de la herencia transgeneracional epigenética inducida por el medio ambiente ha sido bien establecido y tiene un impacto significativo en la etiología de la enfermedad [2, 3] y otras áreas de la biología como la evolución [23]. Aunque la mayoría de las investigaciones anteriores se han centrado en un proceso epigenético individual como la metilación del ADN [3, 4, 10] o el ncRNA [6, 8], pocos han examinado múltiples procesos. Nuestros estudios anteriores demostraron en la herencia de patología transgeneracional epigenética inducida por vinclozolina y DDT que los espermatozoides de la generación F3 transgeneracional tenían regiones de metilación diferencial de ADN (DMR) coordinadamente alteradas, expresión de ARN no codificantes (ncRNA), sitios de retención de histonas diferenciales (DHR) y modificaciones de histonas [24, 25]. Estas observaciones sugieren interacciones potenciales entre los diferentes procesos epigenéticos, pero esto queda por dilucidar durante el fenómeno de la herencia epigenética. Estudios previos han demostrado un papel del ncRNA en la metilación del DNA dirigida por RNA en varios sistemas diferentes [26-28]. El ncRNA puede ayudar a localizar el sitio de metilación del ADN y facilitar los procesos posteriores de remodelación de la cromatina. Por tanto, se ha establecido la integración de la metilación del ncRNA y del DNA. Las modificaciones de las histonas también pueden modificarse drásticamente mediante la remodelación del ARNc y la cromatina para pasar de los sitios de expresión génica activos con eucromatina a los sitios de ADN inactivos con heterocromatina [29]. Aunque se dispone de información sobre la retención de histonas en los espermatozoides y su impacto en el embrión [30, 31], no se ha informado sobre el papel potencial de diferentes procesos epigenéticos en la retención de histonas. Recientemente, se ha observado un papel de las exposiciones ambientales (p. Ej., Vinclozolina y DDT) para promover la herencia epigenética transgeneracional de la retención de histonas de esperma [24, 25, 32]. El estudio actual investiga la integración potencial de la metilación del ADN, el ncRNA y las alteraciones de las histonas en el fenómeno de la herencia transgeneracional epigenética. Los análisis previos de la expresión concurrente de los procesos epigenéticos entre las generaciones F1, F2 y F3 con un umbral estadístico estricto han demostrado una superposición insignificante entre las diferentes generaciones o entre los procesos epigenéticos [24, 25]. El estudio actual utilizó un análisis de superposición extendido con un umbral estadístico menos estricto y encontró superposiciones entre las generaciones y las marcas epigenéticas. Se identificó la potencial integración de los diferentes procesos epigenéticos y la conservación generacional. RESULTADOS El diseño experimental involucró a ratas hembra Sprague Dawley gestantes de la generación F0 a los 120 días de edad expuestas durante los días embrionarios 8-14 (E8-E14) de manera transitoria a vinclozolina (100 mg / kg de peso corporal / día) o DDT (25 mg / kg de peso corporal / día), o control de dimetilsulfóxido de vehículo (DMSO), como se describió anteriormente [24, 25]. La descendencia de la generación F1 se obtuvo y se envejeció hasta los 90 días de edad y luego se crió dentro del linaje (control, vinclozolina o DDT) para generar la gran descendencia de la generación F2. Posteriormente, la generación F2 se crió de manera similar para generar la gran descendencia transgeneracional F3 dentro del linaje. En cada generación o linaje no se utilizó la cría de hermanos o primos para evitar cualquier artefacto de consanguinidad [1, 3]. El sesgo de la camada se evitó sacrificando las camadas a 10 (aproximadamente 5 hembras y 5 machos), y luego solo uno o dos machos y hembras de cada camada se utilizaron para la reproducción dentro del linaje, como se describió anteriormente. Todos los machos tenían una edad de hasta 120 días y se sacrificaban para la recolección de espermatozoides para el análisis molecular, como se describe en estudios previos [24, 25]. El número de animales individuales investigados en cada generación para la recolección de esperma y el análisis molecular fue de aproximadamente 10 a 17 machos, por lo que n = 10 a 17 para animales con tres grupos diferentes de 4 a 6 animales para cada generación y análisis de epimutación. Los espermatozoides recolectados se utilizaron para aislar ARN, ADN y cromatina para el análisis de ncRNA, metilación del ADN, retención de histonas y modificación de histonas, como se describe en estudios previos [24, 25], (Fig. 1). Los datos moleculares de estos estudios previos (GEO # GSE109775, GSE106125 y NCIB SRA: PRJNA430483 largeRNA (control y DTT), PRJNA430740 smallRNA) se analizaron para explorar los datos más bioinformáticamente. 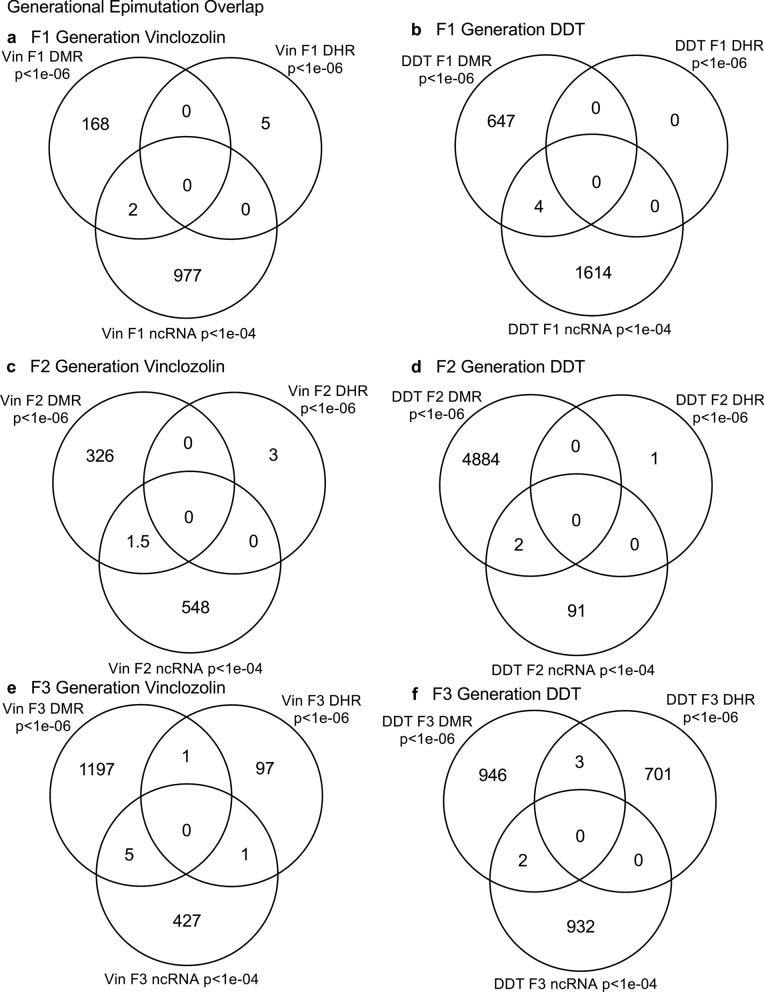 Fig. 1 Generational epimutation overlap at high stringent statistical threshold. a F1 generation vinclozolin lineage DMR (p < 1e−06), DHR (p < 1e−06), and ncRNA (p < 1e−04). b F1 generation DDT lineage DMR (p < 1e−06), DHR (p < 1e−06), and ncRNA (p < 1e−04). c F2 generation vinclozolin lineage DMR (p < 1e−06), DHR (p < 1e−06), and ncRNA (p < 1e−04). d F2 generation DDT lineage DMR (p < 1e−06), DHR (p < 1e−06), and ncRNA (p < 1e−04). e F3 generation vinclozolin lineage DMR (p < 1e−06), DHR (p < 1e−06), and ncRNA (p < 1e−04). f F3 generation DDT lineage DMR (p < 1e−06), DHR (p < 1e−06), and ncRNA (p < 1e−04) Los espermatozoides DMR, ncRNA (tanto sncRNA pequeños como grandes lncRNA) y DHR se analizaron en cada muestra, como se describió anteriormente [24, 25], para las muestras de esperma de la generación de vinclozolina y DDT F1, F2 y F3. Los números y superposiciones de DMR, ncRNA y DHR para cada generación con un umbral de rigurosidad alta se presentan como se informó anteriormente en (Fig. 1). El solapamiento con un diagrama de Venn para la generación F3 transgeneracional para las diferentes marcas epigenéticas es insignificante con el umbral de rigurosidad alta, (Fig. 1e, f), para cada exposición, como se identificó previamente [24, 25]. Las generaciones F1 y F2 también fueron principalmente distintas entre las epimutaciones (Fig. 1a-d). Aunque las diferentes alteraciones epigenéticas están presentes en cada generación para ambos linajes de exposición, las superposiciones con un umbral estadístico estricto fueron insignificantes, lo que sugiere funciones distintas y una falta de integración, como se sugirió anteriormente [24, 25]. Curiosamente, cuando se hizo una comparación de una epimutación con un alto grado de rigurosidad con las otras en p <0,05, se identificaron varias ubicaciones genómicas con los diferentes tipos de epimutaciones presentes. Las ubicaciones cromosómicas de estas marcas epigenéticas alteradas (es decir, epimutaciones) se presentan en (Fig. 2) y en (Archivo adicional 1: Tablas S1-S6) para cada generación para muestras de esperma de linaje de vinclozolina y DDT. Las etiquetas codificadas por colores identifican DMR, ncRNA y DHR en todos los genomas con ubicaciones cromosómicas comunes para cada generación. Solo se muestran aquellos sitios significativos en alta rigurosidad (índice de código de color) con un análisis de epimutaciones que se superponen con las otras epimutaciones en p <0.05, (Fig. 2). En el (Archivo adicional 1: Tablas S1-S6) se presentan las ubicaciones cromosómicas de epimutación específicas, los valores p estadísticos y las asociaciones de genes. En las generaciones F1 y F2 sólo se encontraron alteraciones en los ncRNAs y DMRs, como se describió previamente [24, 25]. Por tanto, las superposiciones se produjeron principalmente entre el ncRNA y DMR en las generaciones F1 y F2 (fig. 2a-d). Las DHR se desarrollaron en la generación F3 transgeneracional, como se describió anteriormente [24, 25]. En las generaciones F1 y F2, el ncRNA fue predominantemente la epimutación estadísticamente significativa alta y la superposición con DMR en el p <0.05, (Fig.2a-c), con una mezcla de ncRNA y DMR en la generación F2 del linaje DDT, (Fig. 2d). La generación F3 transgeneracional también tenía una mezcla de ncRNA y DMR con una significación estadística alta, así como una serie de DHR (Fig. 2e, f). Por lo tanto, las ubicaciones cromosómicas con múltiples epimutaciones se identifican con ncRNA predominando en las generaciones F1 y F2 con el umbral estadístico alto, y las DMR siendo más predominantes en la generación F3 con una mezcla de las diversas epimutaciones (Fig.2 y archivo adicional 1 : Tablas S1-S6). 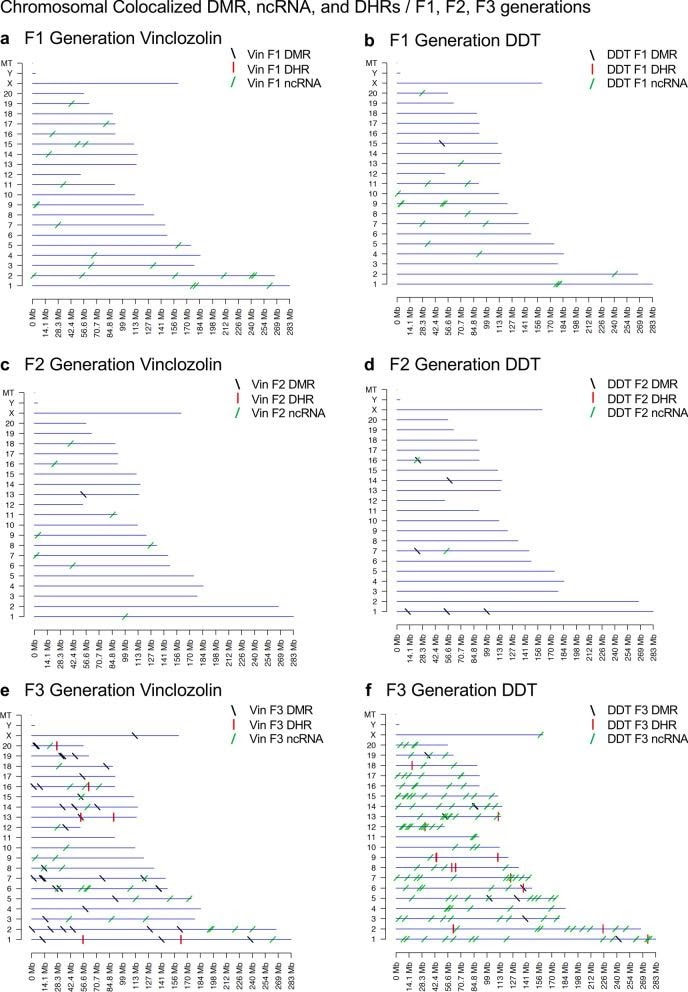 Fig. 2 Chromosomal colocalization of overlap epimutations. The overlap of one epimutation at high statistical stringency (DMR p < 1e−06, DHR p < 1e−06, or ncRNA p < 1e−04) overlap with others at p < 0.05. The epimutation at high stringency is identified with color and marked as indicated by the inset legend. The chromosomal number and size (megabase) are presented. a F1 generation vinclozolin lineage ncRNA and DMR. b F1 generation DDT lineage ncRNA and DMR. c F2 generation vinclozolin lineage ncRNA and DMR. d F2 generation DDT lineage ncRNA and DMR. e F3 generation vinclozolin lineage DMR, DHR and ncRNA. f F3 generation DDT lineage DMR, DHR and ncRNA Se realizó un análisis de superposición extendido con los datos del linaje de DDT y vinclozolin utilizando un umbral estadístico menos estricto para las comparaciones (Fig. 3). Los conjuntos de datos epigenéticos de umbral estadístico más estrictos (DMR p <1e-06, ncRNA p <1e-04 y DHR p <1e-06) se compararon entre las generaciones y las marcas epigenéticas con un umbral estadístico p <0,05. Esto optimizó el potencial para identificar superposiciones en comparación con los umbrales más estrictos utilizados en (Fig. 1). Las filas presentan los umbrales DMR, ncRNA y DHR más estrictos para las generaciones F1, F2 y F3. Las columnas presentan las correspondientes superposiciones de umbral p <0,05 con los conjuntos de datos de umbral de valor p más alto. El examen de las filas horizontales, como se esperaba, muestra una superposición del 100% (es decir, sombreada) para el mismo conjunto de datos y el número de marcas epigenéticas asociadas y el porcentaje (%) de superposición con el valor del margen izquierdo. Esta superposición extendida permite comparar los dos umbrales de rigurosidad diferentes y determinar observaciones adicionales de superposición (Fig. 3). Se observan tendencias similares en las superposiciones para los conjuntos de datos de DDT y vinclozolin. Una de las observaciones iniciales fue que el ncRNA de la generación F1 tenía un alto porcentaje de superposición con el DMR de la generación F3 (Fig. 3 y archivo adicional 1: Tablas S1 y S2). Se hacen observaciones similares con las generaciones F1 y F2. Para la generación F1, el ncRNA de DDT tenía más de un 20% de superposición observado con los DMR de generación F1, F2 y F3, mientras que el ncRNA de vinclozolina F1 tenía aproximadamente un 35% de superposición con los DMR de generación F1, F2 y F3 (Figs. Figs. 33 y y 4a, 4a, b). Las listas de sitios de ncRNA y DMR superpuestos se presentan en (Archivo adicional 1: Tablas S1 y S2). Los ncRNA de generación F2 y F3 fueron similares con la superposición con los DMR de generación DDT de aproximadamente un 20%, pero se redujo a un 10-15% con los DMR de vinclozolina, (Fig. 3). Por lo tanto, algunos ncRNAs eran comunes entre las generaciones y tenían una superposición con los DMR que variaban entre el 8 y el 35% de superposición para los DMR de vinclozolin y 15-20% de superposición para DMR de DDT. Se sugiere la posibilidad de que el ncRNA pueda promover la metilación del ADN dirigida por RNA. Los diagramas de Venn presentados en (Fig. 4a, b) apoyan esas superposiciones y las superposiciones epigenéticas de ncRNA y DMR se enumeran en (Archivo adicional 1: Tablas S1 y S2). 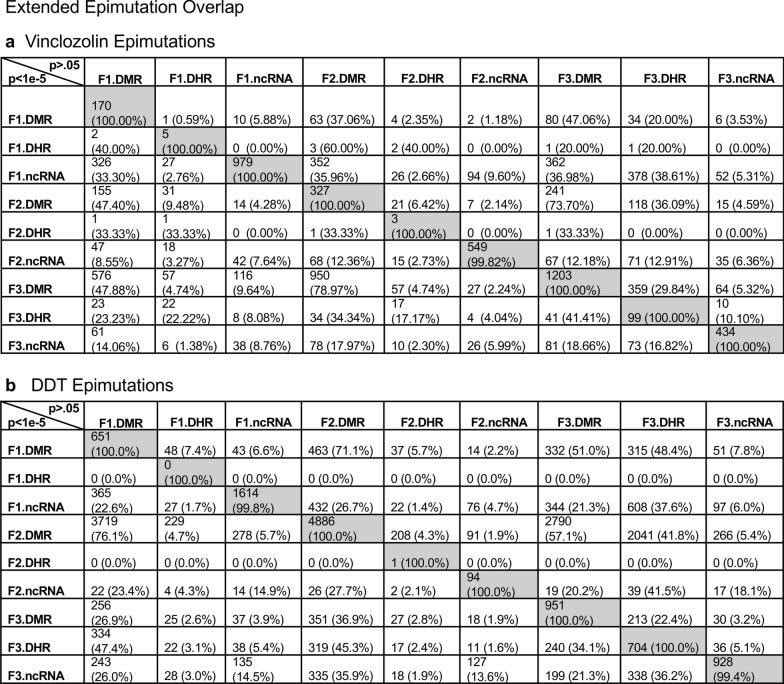 Fig. 3 Extended epimutation overlap. The epimutations at high stringency (DMR p < 1e−06, DHR p < 1e−06, and ncRNA p < 1e−04) in rows were compared to epimutations at p < 0.05 in columns. The number of overlap epimutations and percentage of the total are presented for each overlap. As anticipated, 100% overlap was observed for the same generation and epimutation indicated by shaded box. a Vinclozolin lineage epimutation and b DDT lineage epimutation overlap 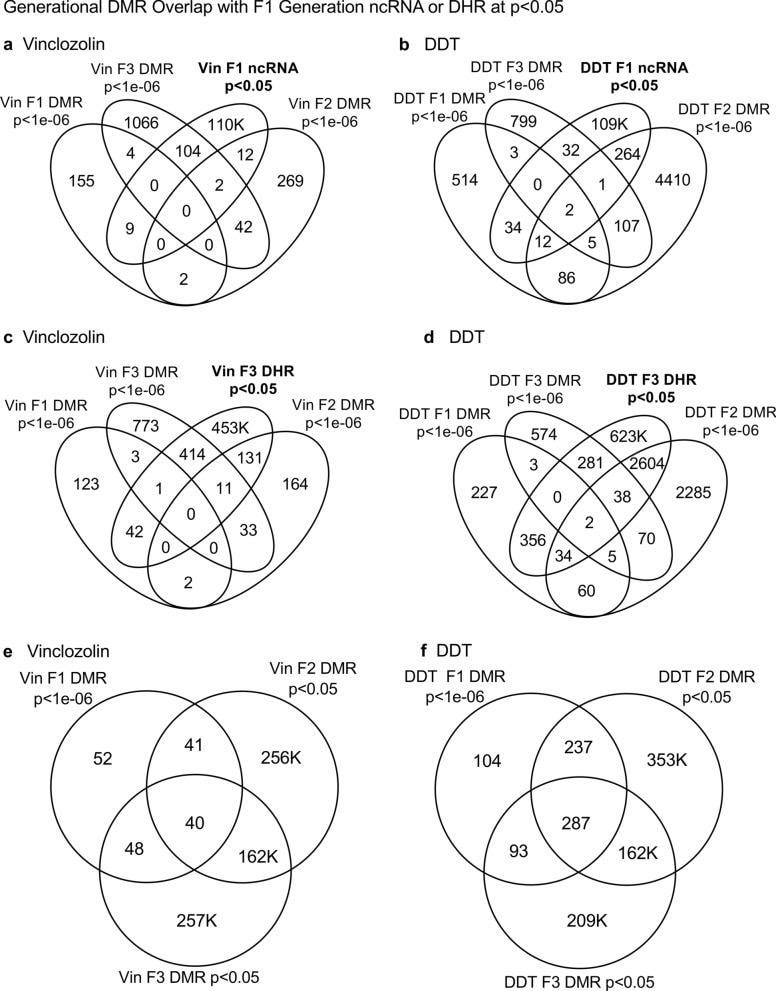 Fig. 4 Epimutation overlaps. Generational DMR overlap with F1 generation ncRNA p < 0.05. A Venn diagram overlap of F1, F2, and F3 generation DMR (p < 1e−06) with F1 generation ncRNA (p < 0.05). a Vinclozolin lineage DMR and ncRNA overlap. b DDT lineage DMR and ncRNA overlap. Generational DMR overlap with F3 generation DHR p < 0.05. A Venn diagram overlap of F1, F2, and F3 generation DMR (p < 1e−06) with F3 generation DHR (p < 0.05). c Vinclozolin lineage DMR and DHR overlap. d DDT lineage DMR and DHR overlap. Generational DMR overlap. A Venn diagram overlap of F1 generation DMR (p < 1e−06) with F2 and F3 generation DMR (p < 0.05). e Vinclozolin lineage DMR overlap. f DDT lineage DMR overlap La siguiente observación fue que los DMR de la generación F1, F2 y F3 tenían una superposición del 20-48% con los DHR de la generación F3 para los linajes DDT y vinclozolin (Fig. 3). Curiosamente, los DHR de la generación F3 tenían una superposición del 23-47% con los DMR de las generaciones F1, F2 y F3 para ambos linajes de exposición. El diagrama de Venn se superpone en (Fig. 4c, d) apoya estas superposiciones de DMR y DHR y sugiere que las DMR pueden ayudar a guiar la formación de DHR transgeneracionalmente. Los DMR y DHR superpuestos de la generación F3 se presentan en (Archivo adicional 1: Tablas S3 y S4). Una observación interesante fue la superposición entre los DMR de la generación F1, F2 y F3 para las exposiciones a DDT y vinclozolina (Figs. (Figs. 33 y 4e, 4e, f). La superposición más alta para los DMR de la generación F1 de DDT fue la F2 DMR de la generación F3 con un 71% de superposición, y para los DMR de vinclozolina F2 con los DMR de la generación F3 con un 73% de superposición. El más alto para los DMR de la generación F3 fue un 79% de superposición con los DMR de vinclozolina F2. Generalmente, un 25-50% Hubo superposición entre los DMR de generación F1, F2 y F3 para ambas exposiciones, (Fig.3). Un diagrama de Venn apoya esta observación y demuestra aproximadamente un 25% de superposición para los DMR de vinclozolina y un 35% de superposición para los DMR de DDT, (Fig. 4e, f). Las listas de estos DMR superpuestos se presentan en (Archivo adicional 1: Tablas S5 y S6). Por lo tanto, un porcentaje (25-35%) de los DMR individuales de la generación F1 se retuvieron transgeneracionalmente. Generalmente, las alteraciones epigenéticas de la generación F3 tenían más superposición entre sí y con las otras generaciones para ambas exposiciones. Se utilizó un análisis de diagrama de Venn para identificar los sitios epigenéticos con DMR, ncRNA y DHR superpuestos (Fig. 4). Los sitios epigenéticos de la generación F3 superpuestos fueron aproximadamente el 25% para los linajes vinclozolin y DDT. Se realizó un análisis de permutación para demostrar que esto es significativamente mayor que la superposición aleatoria observada, con un valor de p de p ≤ 0,05 para los sitios de superposición de 1 kb y 10 kb. Se seleccionaron aleatoriamente varios sitios y se mapearon para identificar las ubicaciones cromosómicas superpuestas de DMR, ncRNA y DHR (Fig. 5). La significación estadística real de las epimutaciones superpuestas en estos ejemplos incluye: (Fig. 5a) (ncRNA p <1e-04, DHR p <0,03 y DMR p <0,005); (Fig. 5b) (ncRNA p <1e-04, DHR p <0,001 y DMR p <0,0004); (Fig. 5c) (DHR p <1e-08, ncRNA p <0,005 y DMR p <0,04); y (Fig. 5d) (ncRNA p <1e-06, DMR p <1e-04 y DHR p <1e-05). En la sección "Discusión" se revisa la posibilidad de que intervenga la metilación del ADN dirigida por ARN y la retención de histonas dirigida por DMR.  Fig. 5 Genomic colocalization of DMR, DHR and ncRNA. The genomic and colocalized DMR, DHR and ncRNA presented. The region size (bp), genes present, and localization of DMR, DHR and ncRNA identified. The various examples include a nc-005100.4, b nc-005104.4, c nc-005111.4, and d nc-005113.4 from the NCBI Rattus norvegicus release 106 in 2016 Todos los análisis previos y superposiciones presentados se basaron en una ubicación cromosómica superpuesta directa para el ncRNA, DMR y DHR. Se abordó la cuestión de si existe un mayor número de sitios con epimutaciones que se encuentran en la misma región pero que no se superponen directamente. Se utilizó una distancia de 5 kb a cada lado de las epimutaciones para tener una ventana de 10 kb para la región de superposición potencial. Se utilizó una superposición extendida utilizando esta ventana de 10 kb con los mismos datos que identificarán los sitios que se superponen directamente y los cercanos dentro de la ventana de 10 kb, Fig. 6. El nivel de superposición con una ventana de 10 kb identificó las mismas superposiciones presentadas y discutidas , pero el nivel de superposición estaba en el rango de 80 a 99% (Fig. 6). Tanto el linaje vinclozolin como el DDT tuvieron el mismo alto nivel de superposición, siendo la mayoría un rango> 90%, con un valor de p de análisis de permutación de p <0,001. Usando esta ventana de 10 kb, la mayoría de ncRNA, DMR y DHR se superpusieron entre las generaciones y las epimutaciones. Esto apoyó todas las observaciones anteriores y demostró un nivel significativo de superposición de epimutación. Dado que aproximadamente el 90% de los DMR de la generación F3 se superpusieron con los DHR de la generación F3 y el ncRNA de la generación F1, los DMR de la generación F3 conservados (archivo adicional 1: Tabla S5 y S6) se utilizaron en un análisis de Pathway Studio para vincular los genes asociados con DMR procesos y patologías celulares, (Archivo adicional 1: S1 y S2). Un gran número de genes asociados con DMR y epimutación vinculados a diversas patologías transgeneracionales previamente observadas, incluyendo enfermedad renal, tumores mamarios, anomalías inmunes, enfermedad de la próstata, enfermedad metabólica o anomalías del comportamiento [3, 33, 34]. 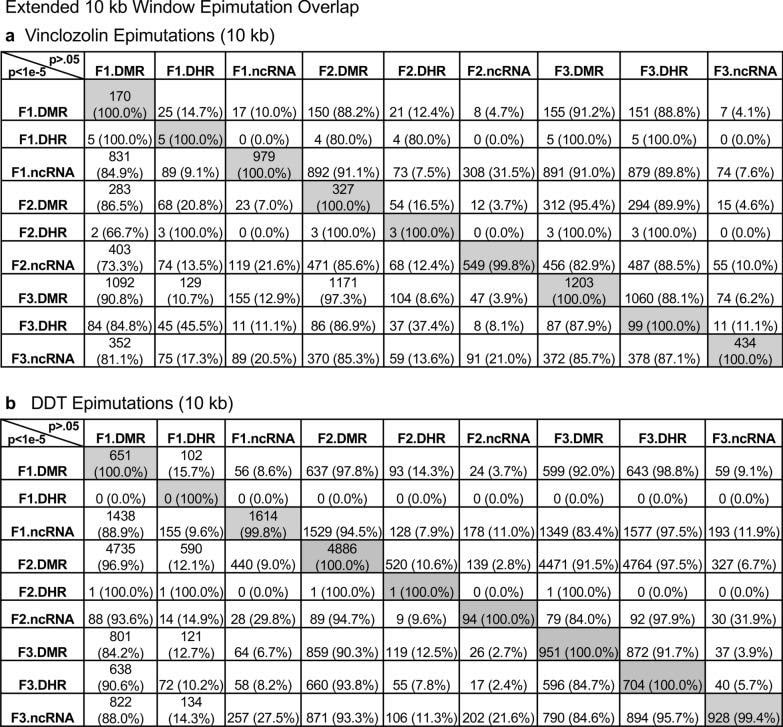 Fig. 6 Extended epimutation overlap within a 10-kb region. The epimutations at high stringency (DMR p < 1e−06, DHR p < 1e−06, and ncRNA p < 1e−04) in rows were compared to epimutations at p < 0.05 in columns. The number of overlap epimutations and percentage of the total are presented for each overlap. As anticipated, 100% overlap was observed for the same generation and epimutation indicated by shaded box. a Vinclozolin lineage epimutation and b DDT lineage epimutation overlap DEBATE Estudios previos han demostrado la presencia concurrente de DMR, ncRNA y DHR en el esperma después de la exposición al DDT o vinclozolina de hembras gestantes de la generación F0 durante la determinación del sexo gonadal [24, 25]. Estos datos se obtuvieron e informaron en una estricta selección de umbral estadístico y demostraron una superposición insignificante en cada generación (Fig. 1) [24, 25]. El estudio actual fue diseñado para investigar más a fondo la integración potencial de los diferentes procesos epigenéticos entre las generaciones F1, F2 y F3. Se adoptó un enfoque para comparar los valores de umbral estadístico más estrictos para DMR, ncRNA y DHR con el umbral p <0,05 menos estricto entre los diferentes procesos epigenéticos y generaciones. Este enfoque de superposición extendido generó una serie de observaciones para sugerir una integración entre generaciones para el fenómeno de herencia transgeneracional epigenética. Una observación interesante de la superposición extendida de DMR demostró que aproximadamente 40-50% de los DMR de espermatozoides de la generación F1 se retuvieron y también estaban presentes en las generaciones F2 y F3 transgeneracional (Figs. (Figs. 33 y 4e, 4e, f). Esto fue 88-97% de los DMR cuando se usaron ventanas de 10 kb, (Fig. 6). Un análisis de permutación demostró que esto era significativo (p <0,001) y no debido a asociaciones aleatorias. La lista de DMR conservados de la generación F1 en las siguientes generaciones se presenta en (Archivo adicional 1: Tablas S5 y S6, y aquellos DMR con asociaciones de genes sugieren que aproximadamente el 50% de estos DMR conservados estaban asociados con genes. Muchos de estos genes tenían asociaciones con una variedad de patologías, Archivo adicional 1: Figuras S1 y S2). Por lo tanto, un porcentaje de los DMR de espermatozoides de la generación F1 se programaron y luego se conservaron en generaciones posteriores. Aunque la mayoría de los DMR de espermatozoides de la generación F1 se conservaron generacionalmente, hubo similitudes mínimas entre en las diferentes generaciones de ncRNAs, (Figs. (Figuras 33 y 6) .6). Las DHR estuvieron presentes principalmente en los espermatozoides de la generación F3, por lo que no se conservaron entre generaciones (Fig.3). En contraste, cuando una región de 10 kb se considera aproximadamente el 40% de las DHR de la generación F3 están presentes en las generaciones F1 y F2, (Figura 6). El papel potencial de estos DMR para la retención de histonas transgeneracional guiada se analiza a continuación. La segunda observación interesante fue la superposición del ncRNA de esperma de generación F1 con los DMR de generación F1, F2 y F3. Más del 20% en DDT y 35% en vinclozolina F1 generación ncRNA se superponen con los DMR de generación F1, F2 y F3, (Figs. (Figs. 33 y y y 44 y archivo adicional 1: Tablas S1 y S2). Las observaciones sugieren el papel potencial de la metilación del ADN dirigida por ARNn en la generación F1 de exposición directa y la generación F3 transgeneracional. La literatura anterior ha establecido un papel para la metilación del ADN dirigida por ARN en varios sistemas biológicos y celulares [26-28]. Esto implica la capacidad del ARNnc para reclutar o dirigir proteínas remodeladoras de la cromatina y proteínas como la metiltransferasa de ADN para guiar la metilación del ADN en un sitio cromosómico, que se ha establecido en una variedad de organismos y procesos de desarrollo diferentes [26-28]. Las observaciones sugieren que la metilación del ADN dirigida por ncRNA puede tienen un papel en el fenómeno de la herencia transgeneracional epigenética. Aunque el ncRNA de la generación F1 tiene la mayor superposición con los DMR de la generación F3, también se observan superposiciones con la F1 y d ARNc de generación F2 con los DMR de varias generaciones, (Fig. 3). Cuando se considera un solapamiento de la región de 10 kb, los ncRNA de la generación F1 tienen un solapamiento del 91% con los DMR de las generaciones F2 y F3, (Fig. 6). Las superposiciones de ncRNA y DMR sugieren que la metilación del ADN dirigida por ncRNA tiene un papel potencial en el proceso de herencia transgeneracional epigenética (Fig. 7). Parece estar implicada una combinación de alteraciones de exposición directa de generación F1 en ncRNA y acciones de generación F3 transgeneracionales posteriores sobre la metilación del ADN. Los sitios epigenéticos colocalizados con ncRNA y metilación del ADN apoyan esta propuesta (Fig. 5). Aunque se ha establecido el proceso molecular de metilación del ADN dirigido por ARN [26-28], y se ha sugerido en los impactos generacionales en plantas y humanos [35, 36], el estudio actual solo demuestra las fuertes correlaciones del ncRNA y los DMR. Se necesitan estudios futuros para proporcionar más conocimientos moleculares y validación de la metilación del ADN dirigida por ncRNA en el fenómeno transgeneracional epigenético. 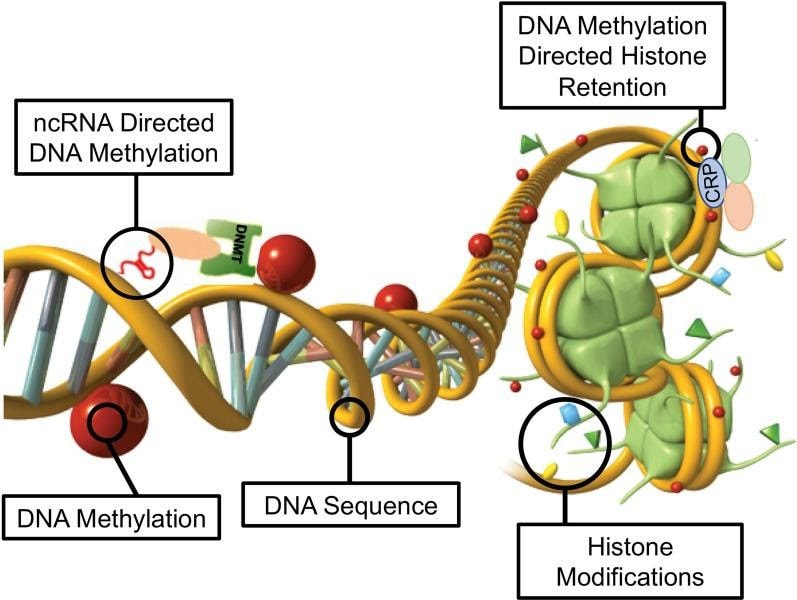 Fig. 7 Diagram of ncRNA-directed DNA methylation and DNA methylation-directed histone retention. The red dot identifies DNA methylation, green histone the nucleosome with modifications in histone tails indicated. The ncRNA association with cofactors and DNA methyltransferase (DNMT) promoting DNA methylation (red dot) for RNA-directed DNA methylation. The DNA methylation (red dot) association with chromatin remodeling proteins (CRP) to promote histone retention is indicated Otra observación interesante fue la superposición de los DMR de la generación F3 transgeneracional con los DHR. Aunque las DHR insignificantes están presentes en las generaciones F1 o F2, la generación F3 tiene DHR que se superponen con las DMR de las generaciones F1 y F2 (Fig. 3). Para los DMR de DDT hubo una superposición de 35 a 50% y para los DMR de vinclozolina, una superposición de 23 a 41%. Considerando una superposición de regiones de 10 kb, las DHR de la generación F3 tenían una superposición del 85-95% con las DMR en todas las generaciones (Fig. 6). El análisis de permutación demostró que este número de superposiciones de regiones de 10 kb no se debe a asociaciones aleatorias (p <0,001). La bibliografía sobre el intercambio espermátido de histonas por protaminas para condensar el ADN en la cabeza de los espermatozoides está bien establecida en la mayoría de los organismos investigados [37-39]. Aunque la gran mayoría del ADN del esperma tiene protaminas asociadas, se retiene un porcentaje de las histonas, que varía entre el 5 y el 10% del ADN en diferentes especies de mamíferos [40]. Anteriormente, encontramos que la retención de histonas se incrementó significativamente en los espermatozoides de la generación F3 transgeneracional con la presencia de nuevos sitios de retención [24, 25, 32]. Por lo tanto, un mecanismo epigenético adicional influenciado durante el proceso de herencia transgeneracional epigenética implica la retención de histonas alterada [32]. La literatura anterior ha descrito las proteínas de transición y los procesos de sustitución de histonas por protaminas [41, 42], pero no se ha considerado el papel de los procesos epigenéticos como la metilación del ADN. Nuestras observaciones anteriores sugieren un papel de este proceso en la herencia epigenética [24, 25]. El estudio actual indica un papel potencial de la metilación del ADN en guiar o dirigir la retención de histonas (Figs. Retención, (Figs. 33 y 6) .6). Estudios anteriores han demostrado un papel fundamental de la metilación del ADN en las acciones de las proteínas remodeladoras de la cromatina [41-43]. Entonces, la metilación del ADN podría alterar las proteínas asociadas y la estructura secundaria del ADN que es un aspecto del proceso de retención de histonas. Aunque se requiere más investigación de los procesos moleculares en estudios futuros, las observaciones del estudio actual sugieren un papel potencial de la retención diferencial de histonas dirigida por metilación del ADN (Fig. 7). Se propone que los DMR ayuden a guiar o dirigir los sitios de retención de histonas de manera que aparezca un mayor número de sitios de forma transgeneracional. Por lo tanto, se propone la existencia de retención de histonas dirigida por metilación del ADN, y las observaciones apoyan una integración de DMR y DHR transgeneracionalmente. Una observación adicional interesante es que los DMR de la generación F1 y F2 que se desarrollan después de la exposición directa a sustancias tóxicas son similares a los DMR de la generación F3, pero que los DHR no se formaron hasta la generación F3 transgeneracional (Figuras 33 y 66).
Los resultados del estudio actual ayudan a integrar los datos anteriores obtenidos con ncRNA, DMR y DHR [24, 25]. Se sugieren los roles potenciales de los DMR dirigidos por ncRNA y los DHR dirigidos por DMR. Un porcentaje de los DMR de la generación F1 se retiene y conserva para las generaciones F2 y F3 posteriores. El ncRNA de generación F1 se superpuso con los DMR de generación F2 y F3, apoyando el papel de la metilación del ADN dirigida por ncRNA y la formación de DMR (Fig. 7). Los subtipos específicos de sncRNA e lncRNA en este proceso requerirán más investigación. Se sugiere el potencial de los DHR dirigidos por DMR, pero se requiere más información para dilucidar los procesos específicos involucrados. Aproximadamente la mitad de las epimutaciones superpuestas tenían genes conocidos asociados. Muchos de estos genes están asociados con patologías previamente identificadas (archivo adicional 1: Figuras S1 y S2), por lo que apoyan un mecanismo de patología transgeneracional. El modelo propuesto y la integración de los ncRNA, DMR y DHR transgeneracionales se presentan en la (Fig. 7). Las observaciones del estudio actual sugieren la integración de procesos epigenéticos en el fenómeno transgeneracional epigenético. Se proporcionan conocimientos sobre el desarrollo y la transmisión generacional de estas epimutaciones de espermatozoides inducidas por el medio ambiente que previamente se ha demostrado que se asocian con el desarrollo y la etiología de la enfermedad. El uso potencial de estos sitios cromosómicos epigenéticos integrados como biomarcadores para identificar la exposición y / o la susceptibilidad a enfermedades sugiere que podrían usarse como diagnósticos para facilitar la medicina preventiva en el futuro. Se necesita más investigación para establecer más a fondo estos mecanismos en el fenómeno de la herencia transgeneracional epigenética, pero el estudio actual proporciona apoyo y un marco para la integración de los diversos procesos epigenéticos. CONCLUSION Las observaciones con las dos exposiciones diferentes de DDT o vinclozolina sugieren que los impactos generacionales y la integración transgeneracional del ncRNA, DMR y DHR son similares. Se observa una variación en el porcentaje de superposiciones, pero las mismas tendencias y conclusiones de integración de las diversas epimutaciones son similares para los linajes de exposición al DDT y vinclozolina. Los sitios de epimutación colocalizados para las diferentes exposiciones demuestran el mismo fenómeno, pero se observan sitios independientes para cada exposición. Los dos modelos diferentes de herencia transgeneracional epigenética inducida por el medio ambiente respaldan el mecanismo general propuesto para la metilación del ADN dirigida por ncRNA y el desarrollo de DHR dirigido por DMR. Aunque el estudio actual identifica tales sitios de epimutación colocalizados e interactuando, muchos de los ncRNA, DMR y DHR específicos no están colocalizados [24, 25]. Por tanto, las acciones independientes de los ncRNA, DMR y DHR también serán importantes en el mecanismo implicado en la herencia transgeneracional epigenética inducida por el medio ambiente. Una combinación de epimutaciones de ncRNA, DMR y DHR desarrolladas durante la gametogénesis permite impactos embrionarios posteriores a la fertilización y sugiere que la integración de ncRNA y DMR estará involucrada en la herencia epigenética. El mecanismo propuesto en (Fig. 7) ayuda a dilucidar los mecanismos moleculares involucrados en el fenómeno de herencia transgeneracional epigenética. RESUMEN DE MATERIALES Y MÉTODOS Estudios y cría de animales Como se describió anteriormente [24, 25] y se amplió en el (Archivo adicional 1: Métodos suplementarios), las ratas macho y hembra Sprague Dawley SD exógenas fueron alimentadas con una dieta estándar con agua ad lib y se aparearon. Las ratas hembras gestantes se expusieron al DDT o vinclozolin, y las crías se criaron dentro de cada linaje durante tres generaciones en ausencia de exposición. La generación F3 se envejeció hasta 120 días para el aislamiento de esperma y el análisis molecular, como se describe en el (Archivo adicional 1: Métodos suplementarios). Los espermatozoides se aislaron y se utilizaron para el análisis epigenético, como se describe en el (Archivo adicional 1: Métodos complementarios). Todos los protocolos experimentales para los procedimientos con ratas fueron aprobados previamente por el Comité de Uso y Cuidado de Animales de la Universidad del Estado de Washington (protocolo IACUC # 6252), y todos los métodos se realizaron de acuerdo con las pautas y regulaciones relevantes. Análisis epigenético, estadística y bioinformática Como se describió anteriormente [44], el ADN se aisló de los espermatozoides recogidos en el momento de la disección. El protocolo de aislamiento de ADN se ha descrito anteriormente [33, 34], (archivo adicional 1: métodos complementarios). Se realizó inmunoprecipitación de ADN metilado (MeDIP), seguida de secuenciación de próxima generación (MeDIP-Seq) en el ADN aislado. Se realizaron MeDIP-Seq, bibliotecas de secuenciación, secuenciación de próxima generación y análisis bioinformático, como se describió anteriormente [33, 34] y en el (Archivo adicional 1: Métodos suplementarios). Todos los datos moleculares se han depositado en la base de datos pública en NCBI (GEO # GSE109775 y GSE106125), y las herramientas computacionales de código R están disponibles en GitHub (https://github.com/skinnerlab/MeDIP-seq) y https: // skinner .wsu.edu / archivos-de-datos-genómicos-y-código-r /. Desde mediados de mayo de 2020, los CDC han estado rastreando informes de casos de síndrome inflamatorio multisistémico en niños (MIS-C), una afección rara pero grave asociada con COVID-19. Los CDC están trabajando para aprender más sobre por qué algunos niños y adolescentes desarrollan MIS-C después de tener COVID-19 o de tener contacto con alguien con COVID-19, mientras que otros no. A partir del 1 de octubre de 2020, el número de pacientes que cumplían con la definición de caso de MIS-C en los Estados Unidos superó los 1,000. En 2021, este número superó los 2.000 al 1 de febrero y los 3.000 al 1 de abril. Última actualización con casos notificados a los CDC el 3 de mayo de 2021 o antes *: * Se están investigando pacientes adicionales. Después de la revisión de datos clínicos adicionales, los pacientes pueden ser excluidos si existen diagnósticos alternativos que explican su enfermedad.  Características de los pacientes con MIS-C que están siendo monitoreados de cerca por CDC por raza y etnia, sexo y edad. Hasta la fecha, la mayoría de los pacientes de MIS-C han sido de raza / etnia hispana / latina o negra no hispana. Las poblaciones hispanas / latinas y negras no hispanas también se han visto afectadas de manera desproporcionada por el COVID-19 en general. Se necesitan estudios adicionales de MIS-C para saber por qué ciertos grupos raciales o étnicos pueden verse afectados de manera desproporcionada y para comprender los factores de riesgo de esta enfermedad.  Próximos pasos MIS-C puede ocurrir semanas después de COVID-19 e incluso si el niño o la familia no sabían que el niño tenía COVID-19. Los CDC y las autoridades estatales estarán monitoreando casos adicionales y adaptarán las recomendaciones de MIS-C según sea necesario. Los investigadores están evaluando los casos notificados de MIS-C y los resultados de salud asociados para tratar de aprender más sobre los factores de riesgo específicos de MIS-C, la progresión de la enfermedad en niños y adolescentes, y cómo identificar mejor MIS-C y distinguirlo de otros similares. enfermedades. Sobre los datos Esta información fue tomada de https://www.cdc.gov/mis-c/cases/index.html, la misma que es actualizada el primer viernes de cada mes. Comentarios adicionales Algunos pacientes pueden cumplir con los criterios totales o parciales para la enfermedad de Kawasaki, pero se deben informar si cumplen con la definición de caso de MIS-C Considere MIS-C en cualquier muerte pediátrica con evidencia de infección por SARS-CoV-2 Momento de la presentación de informes La notificación de casos puede retrasarse debido a la capacidad limitada en los departamentos de salud locales / estatales y a medida que los CDC evalúan los datos para garantizar que los casos cumplan con la definición de caso de MIS-C. Fuente: https://www.cdc.gov/mis-c/cases/index.html |
FUCOBISomos una organización al servicio de la salud ambiental y poblacional trabajando por la conservación y recuperación de nuestros recursos naturales a lo largo plazo en defensa de la salud humana. Categoríasarchivo
Octubre 2022
|
- CONÓCENOS
- English
- Premios
-
UNA SALUD
- Proyectos para Estudiantes
- mangroveENCODE >
- shrimpENCODE >
-
childrenENCODE
>
- Proyecto Foldscopes: From Southborough to Ecuador >
- Shrimp Scampi
- Somos lo que Comemos
- Malformaciones Congénitas (Metales y COPs)
- Cancer y EDCs (metales y COPs)
- Aprenda sobre los Contaminantes que afectan a las Personas
- Genes que causan Alergias en Humanos
- Resistencia a Antibióticos y la Industria Avícola
- Contacto
- Eventos
- Publicaciones
- Como puedes ayudar
- Blog
 Canal RSS
Canal RSS
